Malattie degli occhi casi clinici reali
Descrizione di casi clinici reali correlati con iconografia originale, diagnosticati dal titolare del sito, dottor Amedeo Lucente, particolarmente interessanti e clinicamente rilevanti, con considerazioni personali e spesso innovative. Il sito si riserva l'esclusiva originale titolaritá; dell'iconografia e la proprietá intellettuale dei contenuti a fini scientifici e divulgativi. Tutti i contenuti di queste pagine sono di proprietá; esclusiva del sito amedeolucente.it Ne è vietata la riproduzione, anche parziale, se non dietro richiesta scritta al titolore, che ne consentirà o meno l'utilizzo.
Malattie degli occhi :
Foro Retinico Autosbarrato

Foro retinico periferico spontaneo che si � autochiuso altrettanto spontaneamente in giuvane soggetto senza altre problematiche retiniche. A volte questa possibilit� esiste e posso testimoniarla perch� il soggetto � mio paziente che seguo da anni che aveva precedentemente la stessa patologia senza iperpigmentazione spontanea intorno al piccolo e. per altro, non regmatogeno foro.
Rottura retinica gigante
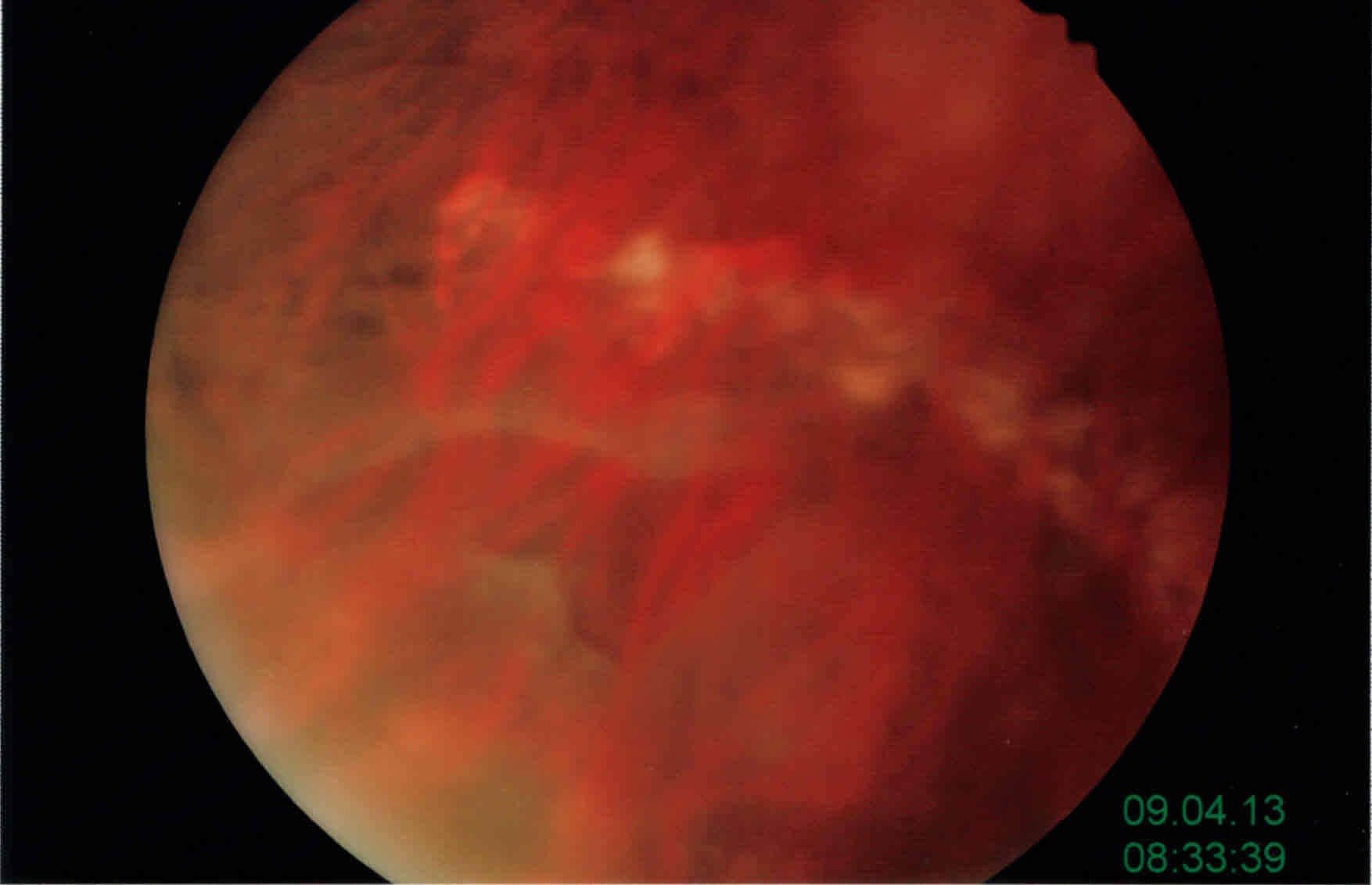
Grande rottura retinica in soggetto giovane miope elevato trattato con successo con argon laser in questo studio. La rottura � stata completamente circoscritta da tre file di spot, nel quadrante infero nasale. Il quadrante infero nasale, meno foriero di distacchi di grandi estensioni, ha aiutato a non far evolvere la rottura. Sicuramente le trazioni vitreali non erano particolarmente tenaci e il trattamento, a distanza di 8 mesi, tiene, senza bisogno di alcun altro intervento; il visus non ha subito menomazioni.
Occlusione parziale Arteria Centrale Retina
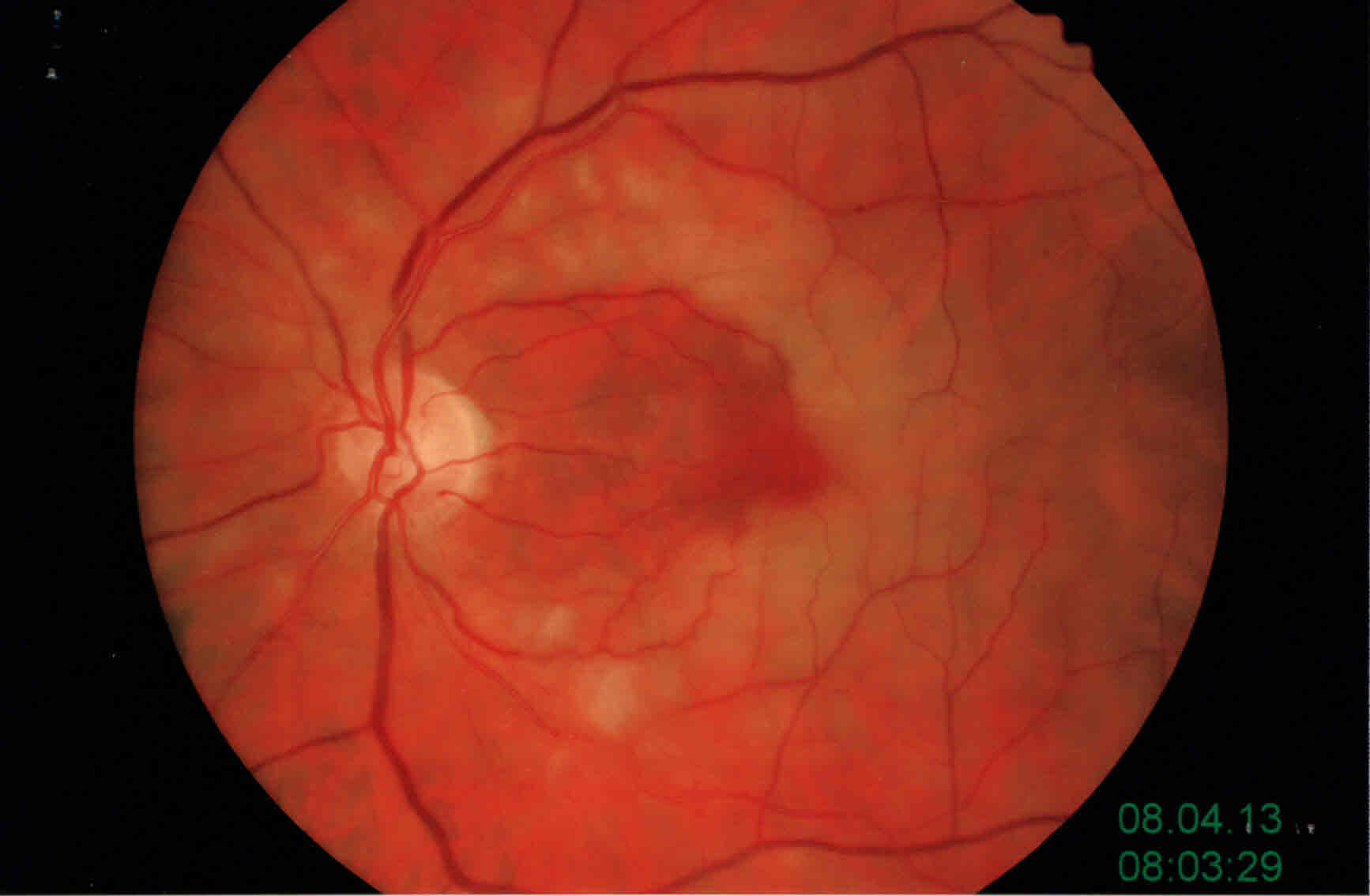
Quadro retinico di occlusione parziale dell'arteria centrale della retina con edema retinico e risparmio dell'area maculare.
Vitreopatia Asteroide
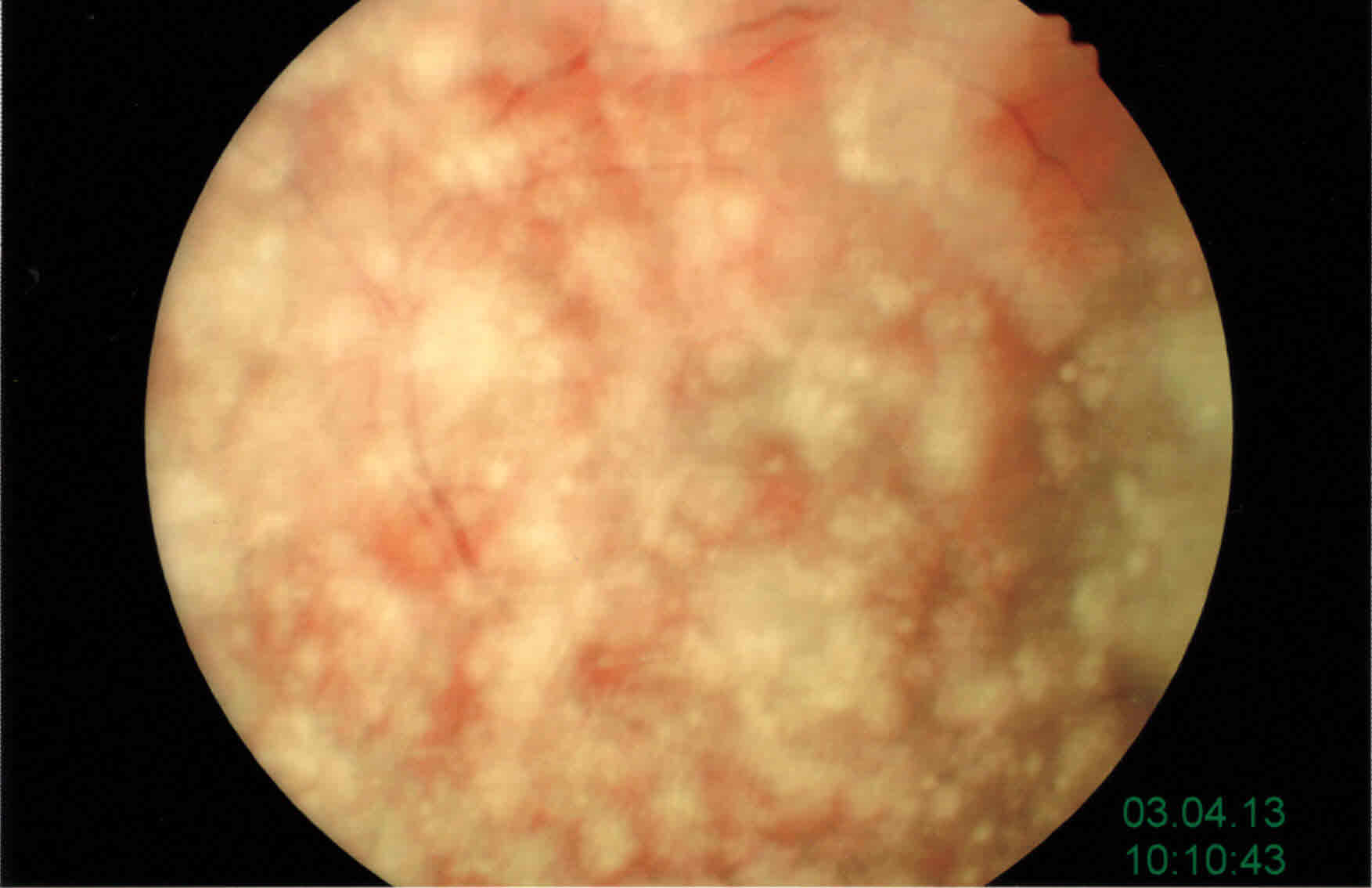
Caso di Vitreoparia asteroide visitato in questo studio che differisce dalla sinchisi scintillante anche per non permettere un visus ottimale. Di seguito la dattagliata descrizione delle vitropatie fatta dall'ottimo maestro d'oftalmologia Raffaello di Lauro e la sua equipe. Varie condizioni degenerative possono interessare il corpo vitreo e la retina, anche se l�intima funzione di queste due strutture crea spesso difficolt� a determinare quale tessuto sia stato interessato per primo nel processo patologico. Con il termine di �degenerazioni vitreo-retiniche� sono raggruppate quelle situazioni patologiche in cui le alterazioni coinvolgono entrambe le strutture. Le degenerazioni vitreo-retiniche possono essere di origine ereditaria �vitreoretinopatie ereditarie� (Tab. I) o acquisite �ialoidoretinopatie�.Queste ultime (Tab. II) possono essere sia legate all�invecchiamento del vitreo (sineresi vitreale e distacco del vitreo), sia secondarie alla presenza di depositi extracellulari, proteici (amiloidosi vitreale) o lipidici (vitreopatiaasteroide e sinchisi scintillante). Nel presente capitolo vengono discusse tre condizioni cliniche comunemente indicate con il termine di vitreopatie degenerative: - Vitreopatia asteroide - Sinchisi scintillante - Amiloidosi del vitreoIl termine di vitreopatie degenerative � tuttavia improprio (1) dal momento che implica un processo primario a livello del gel vitreale, mentre l�unica degenerazione primaria del vitreo � rappresentata dalla sineresi e dal distacco di vitreo, mentre la maggior parte delle vitreopatie degenerative sono secondarie a degenerazioni retiniche ed il materiale che si accumula nel vitreo proviene da strutture diverse da esso, anche se questo non � ancora stato dimostrato in maniera definitiva nel caso della vitreopatia asteroide.VITREOPATIA ASTEROIDE La degenerazione asteroide del vitreo � stata descritta per la prima volta da Schmidt (2) ed � stata differenziata dalla sinchisi scintillante da Benson nel 1894 (3). Nella letteratura europea si possono ancora incontrare le varianti proposte da Wiegmann (4): �Scintillatio nivea� e �scintillatio albescens�. La vitreopatia asteroide � un�affezione rara, con una prevalenza nella popolazione generale tra 0.15% e 0.9% (5). Unilaterale nel 75% dei casi, si riscontra generalmente nei soggetti d�et� superiore ai 60 anni ed � pi� frequente nei diabetici. Clinica. L�aspetto caratteristico della vitreopatia asteroide � rappresentato dalla presenza di opacit� rotonde bianco-giallastre e brillanti, sospese nel gel vitreale (Fig. 1), spesso aggregate in ammassi e dall�aspetto simile a fili o grappoli di perle.Ph. 1 Vitreopatia asteroideIl vitreo presenta un aspetto normale: la liquefazione vitreale ed il DPV sono presenti solo nel 12% dei soggetti (6). I corpi asteroidi sono attaccati alle fibre del collageno e sono mobili con i movimenti del gel vitreale. All�esame ecografico si osserva una moltitudine di picchi iperecogeni, mobili con il movimento del bulbo oculare. Una caratteristica costante � la presenza di una corona vitreale ipoecogena tra l�immagine iper-riflettente dei corpi asteroidi e la superficie anteriore della retina, che pu� essere confusa con un distacco posteriore del vitreo. La vitreopatia asteroide � generalmente asintomatica. Anche se le opacit� sono talmente dense da ostacolare la visione del fondo oculare, i pazienti non hanno una riduzione dell�acuit� visiva e la malattia rappresenta spesso una scoperta fortuita durante un esame oculistico di routine. Quando � presente una notevole diminuzione visiva, bisogna sospettare una patologia retinica associata (degenerazione maculare legata all�et�, retinopatia diabetica, ecc.). Istopatologia. L�analisi al microscopio elettronico a trasmissione ha evidenziato la presenza di lipidi complessi e l�analisi a diffrazione di raggi X ha mostrato che le particelle asteroidi sono formate dall�apposizione di strati concentrici di fosfolipidi associati a complessi calcio-fosfato, intimamente legati al collageno vitreale (6). Eziologia. L�eziologia resta ancora sconosciuta. Non sono state dimostrate associazioni con altre condizioni patologiche oculari o sistemiche (7): il legame con il diabete mellito, riportato in alcuni studi iniziali (8), � stato successivamente messo in dubbio per carenze metodologiche nelle ricerche effettuate (9). Nessuno studio, inoltre, ha riportato l�associazione di ialopatia asteroide con una dislipidosi ereditaria. Trattamento. La degenerazione asteroide richiede raramente un trattamento terapeutico perch� � di solito asintomatica e scevra di complicanze (10). Nei rari casi in cui � presente una massiva concentrazione di corpi asteroidi nella parte centrale della cavit� vitreale, pu� verificarsi una discreta diminuzione visiva. Una scarsa visibilit� del fondo oculare non consente, talvolta, la sorveglianza di una patologia del segmento posteriore e l�eventuale realizzazione di una fotocoagulazione retinica. In questi casi pu� essere proposta una vitrectomia via pars plana (10-14) (Fig. 2).Ph. 2 Trattamento vitreopatia asteroideTuttavia la vitrectomia nella degenerazione asteroide non � esente da rischi (1, 11, 15) e potrebbe addirittura favorire un distacco di retina postoperatorio. Questa complicanza si verifica probabilmente perch� in questa patologia il distacco posteriore di vitreo e la liquefazione vitreale sono significativamente meno frequenti rispetto alla popolazione normale d�et� analoga (p<0.01), mentre l�aderenza vitreoretinica � maggiore (1).SINCHISI SCINTILLANTE La sinchisi scintillante � una condizione degenerativa del vitreo spesso confusa con la vitreopatia asteroide, dalla quale deve essere differenziata sia sul piano clinico che su quello patologico. Clinica. Descritta per la prima volta da Sichel nel 1846 (16), si presenta clinicamente con minute multiple particelle cristalliniche, brillanti e colorate, sospese in un vitreo degenerato e liquefatto (17). L�aspetto di questi corpuscoli � piatto ed angolare, contrariamente a quello rotondeggiante dei corpi asteroidi. I cristalli sono mobili con i movimenti dell�occhio, ma indipendenti dai movimenti del vitreo, a differenza dei corpi asteroidi che sono strutturalmente associati al vitreo. Durante i movimenti oculari si diffondono rapidamente nella cavit� vitrea come coriandoli multicolori, ma si depositano per gravit� sul fondo oculare, quando l�occhio � immobile. Nei pazienti fachici le particelle possono accumularsi in camera anteriore (Fig. 3) e simulare un ipopion (18, 19). Talvolta si pu� sviluppare un glaucoma secondario (18, 19).Ph. 3 Sinchisi scintillanteL�aspetto ecografico corrisponde ad una moltitudine di punti mediamente ecogeni. La sinchisi scintillante viene riscontrata pi� spesso in soggetti al di sotto dei 35 anni di et� (20), mentre la ialoidopatia asteroide si manifesta negli anziani. La sinchisi scintillante si presenta esclusivamente in occhi gravemente traumatizzati o affetti da infiammazioni croniche (17) o in seguito ad emorragie intravitreali (21). Istopatologia. Gli studi istopatologici hanno ulteriormente differenziato la sinchisi scintillante dalla vitreopatia asteroide. I cristalli della sinchisi, infatti, sono costituiti da cristalli di colesterolo, mentre i corpi asteroidi risultano composti da un complesso calcio-fosfolipidico (22). Per questo motivo il termine pi� appropriato per quest�affezione sarebbe �cholesterosis bulbi�. Eziologia. L�esatta eziologia dei cristalli di colesterolo non � chiara. Con ogni probabilit� il colesterolo deriva dalla distruzione cellulare di globuli rossi o leucociti lisati provenienti da emovitreo o flogosi cronica (22). Trattamento. La presenza di cristalli di colesterolo in camera vitrea non richiede alcun trattamento; tuttavia i pazienti afachici con quest�affezione possono sviluppare un glaucoma, quando le particelle ostruiscono l�angolo di filtrazione. In questi casi � utile praticare una vitrectomia ed un lavaggio della camera anteriore.AMILOIDOSI VITREALE L�amiloidosi � una patologia caratterizzata dalla deposizione extracellulare di una sostanza proteica anomala che presenta caratteristiche tintoriali, biochimiche e ultrastrutturali particolari. La proteina anomala � una prealbumina, dipendente da una mutazione a livello della transtiretina. La patogenesi di questa complessa malattia rimane oscura. La classificazione anatomo-patologica attuale distingue le amiloidosi in sistemiche (poliviscerali) o localizzate (monoviscerali), primarie o secondarie, familiari o idiopatiche (23). Le localizzazioni oculari sono multiple e possono interessare i muscoli oculo-motori, la ghiandola lacrimale, le palpebre, le congiuntive, la cornea, il vitreo, la retina e l�orbita. Nonostante l�amiloidosi come malattia a s� stante sia stata riconosciuta gi� agli inizi del XIX secolo, l�interessamento vitreale � stato osservato solo nel 1953 (24). Clinica. Le alterazioni del vitreo si repertano essenzialmente nelle neuropatie amiloidi eredofamiliari tipo I e II e possono rappresentare la prima manifestazione della malattia, insorgendo molto tempo prima del coinvolgimento di altri organi. Si presentano clinicamente (23, 25, 26, 27) come depositi biancastri a contatto dei vasi retinici, in particolar modo attorno alle arteriole, simulando l�aspetto di un essudato cotonoso. La densit� dei depositi d�amiloide aumenta progressivamente giungendo a coprire interi tratti di vasi retinici. Le opacit� si estendono successivamente nella cavit� vitreale formando aggregati granulari con bordi lievemente sfrangiati che si diffondono in senso postero-anteriore e s�inseriscono alla faccia posteriore del cristallino sotto forma d�opacit� biancastre con pseudopodi, assumendo l�aspetto di �veli di merletto� o di �lana di vetro�. Questo aspetto, bilaterale e simmetrico, � specifico della malattia. L�amiloidosi vitreale � spesso asintomatica. Una diminuzione visiva si verifica quando le opacit� vitreali sono dense o si accumulano a livello della capsula posteriore del cristallino. Raramente � associata a complicanze, tuttavia la sostanza amiloide pu� strozzare e obliterare i vasi coroideali e/o retinici e comportare neovascolarizzazione retinica periferica ed emorragie vitreali (28). Nella forma evolutiva si pu� sviluppare un glaucoma ad angolo aperto secondario alla presenza di depositi amiloidi nel trabecolato. Un glaucoma secondario pu� insorgere anche per un aumento della pressione venosa episclerale in caso di lesione amiloide delle vene episclerali (29). Trattamento. L�unica forma di trattamento dell�amiloidosi vitreale � la vitrectomia via pars plana (26, 30-33). L�intervento � indicato, quando le opacit� vitreali provocano una significativa riduzione visiva. La vitrectomia deve essere completa e soprattutto retrolentale, perch� le recidive sono generalmente dovute alla permanenza di vitreo dietro il cristallino. Se � presente un distacco posteriore del vitreo la prognosi � favorevole. Se invece il vitreo non � distaccato, esiste il rischio di provocare emorragie e rotture retiniche iatrogene e di non poter rimuovere le opacit� amiloidi saldamente aderenti alla retina.Prof. Raffaello di Lauro, Dott.ssa Maria Teresa di Lauro, Dott.ssa Raffaella di Lauro, Dott. Alessandro Senese, Dott.ssa Paola Giustiniani, Dott.ssa Antonietta D�AloiaDivisione Oculistica del CTO � NAPOLI
Morning Glory Anomaly

Bambino di 7 anni non diagnosticato prima e comunque mai fotografato, visitato in questo studio. Dalla letteratura si evince che l'aspetto � assolutamente patognomotico e tra i pi� iconografici, caso clinico che mi hanno richiesto colleghi esteri per loro pubblicazioni. L'altro occhio � normale.Da Wikipedia:Coloboma of optic nerve, also called Morning Glory syndrome, is a rare defect of the optic nerve that causes moderate to severe blindness. It is extremely rare, occurring in only one person per every two million in the United States.Contents [hide] 1 Definition2 Effects2.1 Theories3 Rarity3.1 Diagnosis4 Notable Victims5 See also6 References[edit] DefinitionMorning Glory syndrome is a congenital anomaly of the optic disc in which there is a funnel-shaped excavation of the posterior fundus incorporating the optic nerve, surrounded by an elevated annulus of chorioretinal pigment.[1] The issue stems from an undeveloped optic nerve: while in utero, the nerve ending from the eye never reached the nerve ending from the brain. A central core of white glial tissue occupies the position of the normal optic cup, causing a white mass. When a picture is taken of the eye, this white mass stands out apart from the veins of the eyes, looking very much like the center of a Morning glory flower. Reflection from within the eye may give the appearance of a white pupil, due to the lack of the "black" optic nerve mass, although this "clouded pupil" cannot be used to diagnose the disease. Morning Glory Syndrome typically affects only one eye; but cases has been documented of bilateral Morning Glory Syndrome (those with that stage of severe blindness have other disorders as well, like autism[2]). Also, this syndrome is different from an optic disc injury, as the disorder is present at birth and not developed later in life. Young girls are twice as likely to be diagnosed with this disorder, pointing toward the possibility of a genetic connection. Morning glory syndrome may be associated with Aicardi syndrome, a condition found exclusively in girls (lethal in males).[edit] EffectsVision in the affected eye is severely impaired, depending on the completed development of the optic nerve. More mild cases cause limited depth or color perception, while the most severe cause total blindness. Full use of the affected eye is never gained. The most common side effect of this disorder is eyestrain from overuse of the un-affected eye. Also, there is an increased risk of serious retinal detachment, manifesting in 1/3 of patients. If retinal detachment does occur, it is usually not correctable and all sight is lost in the affected eye.[edit] TheoriesThe exact cause of this syndrome is unknown. This condition may be associated with brain and kidney disorders including papillorenal syndrome, a PAX2 gene mutation. Many times, this syndrome manifests itself alongside other midline or cranial disorders, but it does not seem to be caused by those disorders. Additionally, recent research points toward the possible connection between undiagnosed thyroid issues in the mother during the pregnancy and this syndrome. No conclusive evidence was found, however, due to the rare nature of this disease and its late appearance during childhood (negating the possibility of a neo-natal study).It can be associated with PAX6.[3][edit] RarityMorning Glory Syndrome is extremely rare. High-end guesses claim that this syndrome, first documented and diagnosed in the late 1970s, affects approximately one person out of every million in the United States. More conservative speculation place that number closer to one out of every two or three million. Unfortunately, no surveys have been completed to document the exact effect rate of this syndrome, and very few research articles have been published in the United States on the topic since its first discovery. Several French organizations claim to be knowledgeable on the topic, but the published research seems lacking even from them.[edit] DiagnosisThe first noticeable signs of the syndrome usually do not appear until after the first twelve months of the child's life. The child usually has severe balance issues as he or she learns to sit or walk, often leaning or tilting the head toward the good eye to correct the brain's skewed perception of the world. Often the child will fall in the same direction while walking or run into objects that are placed on his or her blind side. The child will also develop a "lazy eye", or an eye that drifts severely away from the child's fixed point of reference. This syndrome must be confirmed through pupillary dilation and examination of the optic disc, as the signs alone will not constitute a diagnosis.Children with Morning Glory live relatively normal lives, even those who have discolored or lazy pupils. Although non-prescription glasses should be worn for eye protection, this syndrome does not usually prevent the individual from living a normal life, driving cars, playing sports, reading, etc. Certain activities, such as gymnastics and ice skating, may be more difficult for patients with Morning Glory due to a compromised view of the world and skewed sense of balance, but they are still possible. Like most other eye conditions, a diagnosis of Morning Glory precludes a person from certain occupations, such as being a surgeon or fighter pilot.
Loop di IOL sulla sclera
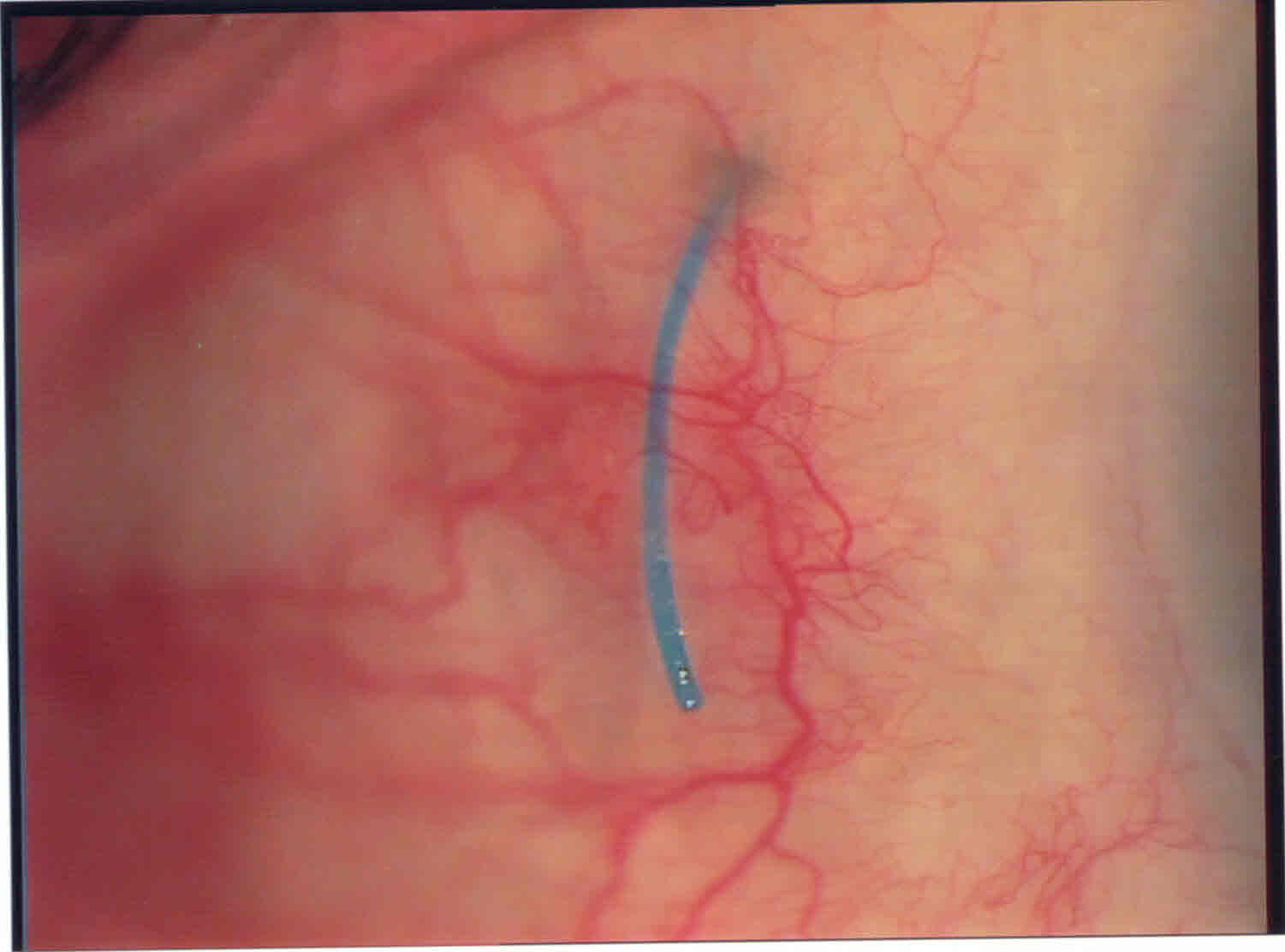
Impianto in camera posteriore di IOL caduta in camera vitrea.