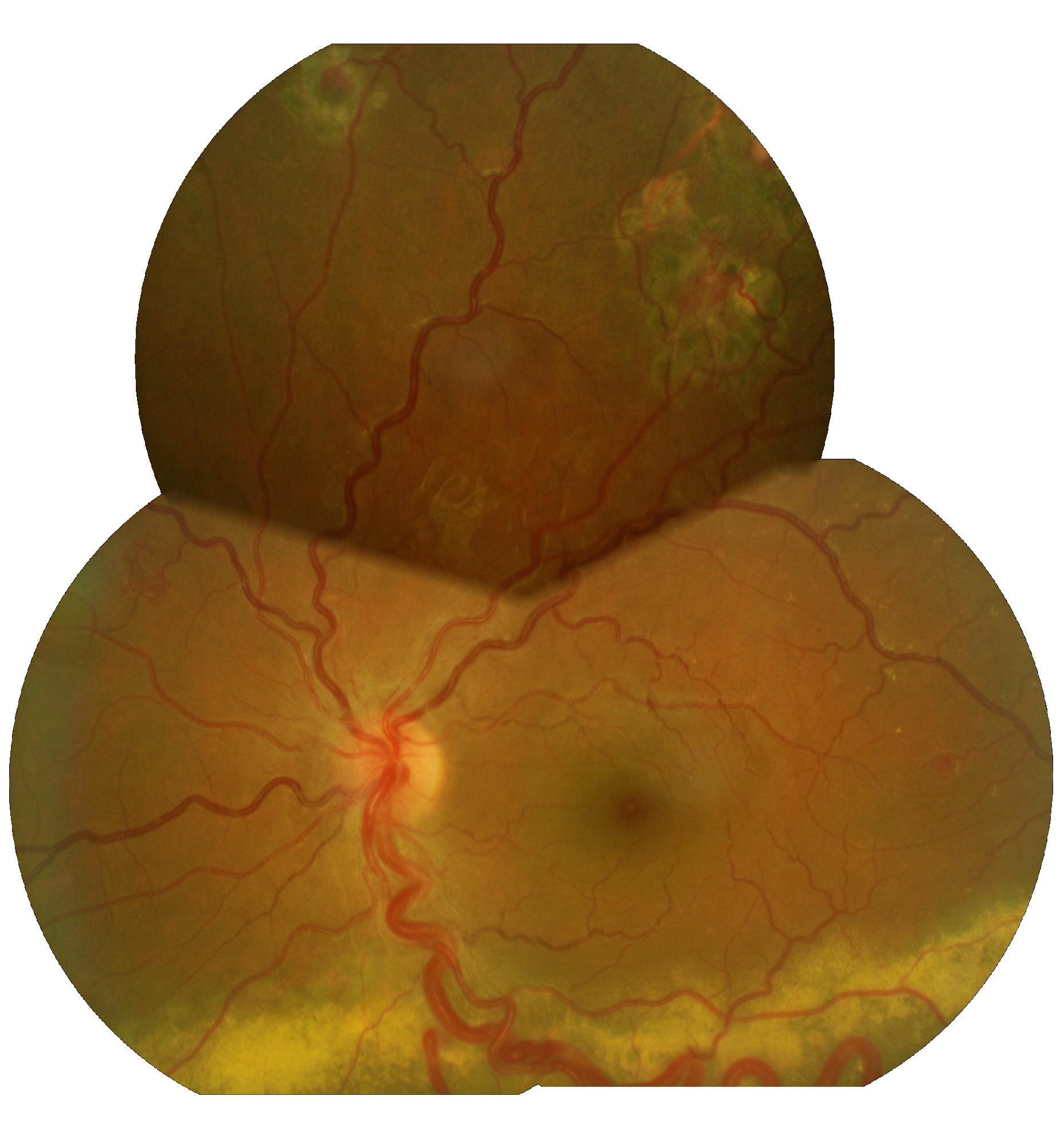
Occhio sinistro maggiormente interessato dalla patologia aneurismatica e dove si evidenzia un'essudazione della retina inferiore con soggettiva diminuzioine del campo visivo.
Sindrome di Von Hippel-Lindau
Da Wikipedia, l'enciclopedia libera
Le informazioni qui riportate hanno solo un fine illustrativo: non costituiscono e non provengono n� da prescrizione n� da consiglio medico. Wikipedia non d� consigli medici: leggi le avvertenze.
Sindrome di Von Hippel-Lindau
La VHL o sindrome di Von Hippel-Lindau � una malattia a carattere ereditario molto rara, caratterizzata dall'associazione di diverse forme di neoplasia, fra cui angiomi e altre forme di neoplasia del rene e feocromocitomi.
Indice
[nascondi] 1 Epidemiologia
2 Cenni storici
3 Eziologia
4 Sintomatologia
5 Diagnosi
6 Terapia
7 Bibliografia
8 Voci correlate
9 Altri progetti
Epidemiologia [modifica]
Colpisce una persona su 36.000. L'incidenza � maggiore intorno ai 30-40 anni, anche se pu� manifestarsi a qualunque et�.
Cenni storici [modifica]
Il nome della sindrome � dovuto a coloro che per primi la descrissero: l�oftalmologo tedesco Eugen Von Hippel e il patologo svedese Arvid Lindau.
Eziologia [modifica]
La causa di questa malattia � una mutazione di un gene localizzato sul braccio corto del terzo cromosoma, mappato in 3p25-26, il quale codifica per la proteina VHL. Quest'ultima, normalmente, legandosi ai fattori di trascrizione HIF (Hypoxia Induced Factor) idrossilati dalla presenza di ossigeno, ne provoca l'ubiquitinizzazione e la distruzione tramite il proteasoma. Il complesso coi geni HIF � responsabile delle risposte cellulari all'ipossia, fra cui lo sviluppo di neoangiogenesi e l'induzione della proliferazione cellulare; quindi VHL si comporta come un oncosoppressore e da questo dato sono facilmente deducibili le conseguenze cliniche della sua mancanza. Il prodotto genico pu� essere totalmente assente (delezioni o mutazioni frameshift) con espressione anomala di codoni di STOP) oppure inattivo, portando a quadri clinici differenti.
Sintomatologia [modifica]
A seconda del genotipo del soggetto sono rinvenibili quadri clinici differenti; in particolare la malattia � suddivisibile in 2 tipi. Il tipo 1 con assenza di feocromocitoma e il tipo 2 che invece lo manifesta. Nel primo caso la proteina spesso � mutata, ma presente; nel secondo caso invece � deleta. Inoltre il tipo 2 � diviso ulteriormente in 3 sottotipi: A, B e C (il primo con tumore a cellule renali, il secondo con anche emangioblastoma e il terzo con solo feocromocitoma). Sono anche riscontrati tumori endocrini (specie pancreatici), cisti renali (dovute all'edema da neoangiogenesi) e altre manifestazioni neoplastiche.
Diagnosi [modifica]
La ricerca delle alterazioni del gene VHL deve essere guidata dalla clinica con cui la patologia si presenta e dalla buona pratica clinica. (bisogna valutare l'utilit� reale e la metodica pi� adatta nel fare una consulenza genetica). In particolare nel caso in cui sospetti una delezione dovr� fare gli esami adatti a trovarla, nel caso in cui avr� invece una mutazione puntiforme user� delle metodiche adatte a cercare queste ultime; fatto ci� nel secondo caso dovr� valutare se la mutazione eventualmente trovata corrisponde a patologia; o tramite la consultazione di database appositi oppure facendo delle prove indirette (cerco nei tumori del soggetto anche la delezione dell'allele sano oltre a quello mutato congenitamente, in modo da provare che il danno trovato � responsabile del quadro clinico)
Terapia [modifica]
Si procede ad intervento chirurgico, che viene effettuato in particolari condizioni:
Lesioni spinali sintomatiche
Lesioni cerebrali sintomatiche o in rapida evoluzione
Neoplasie solide renali > 3 cm
Neoplasie solide pancreatiche > 3 cm
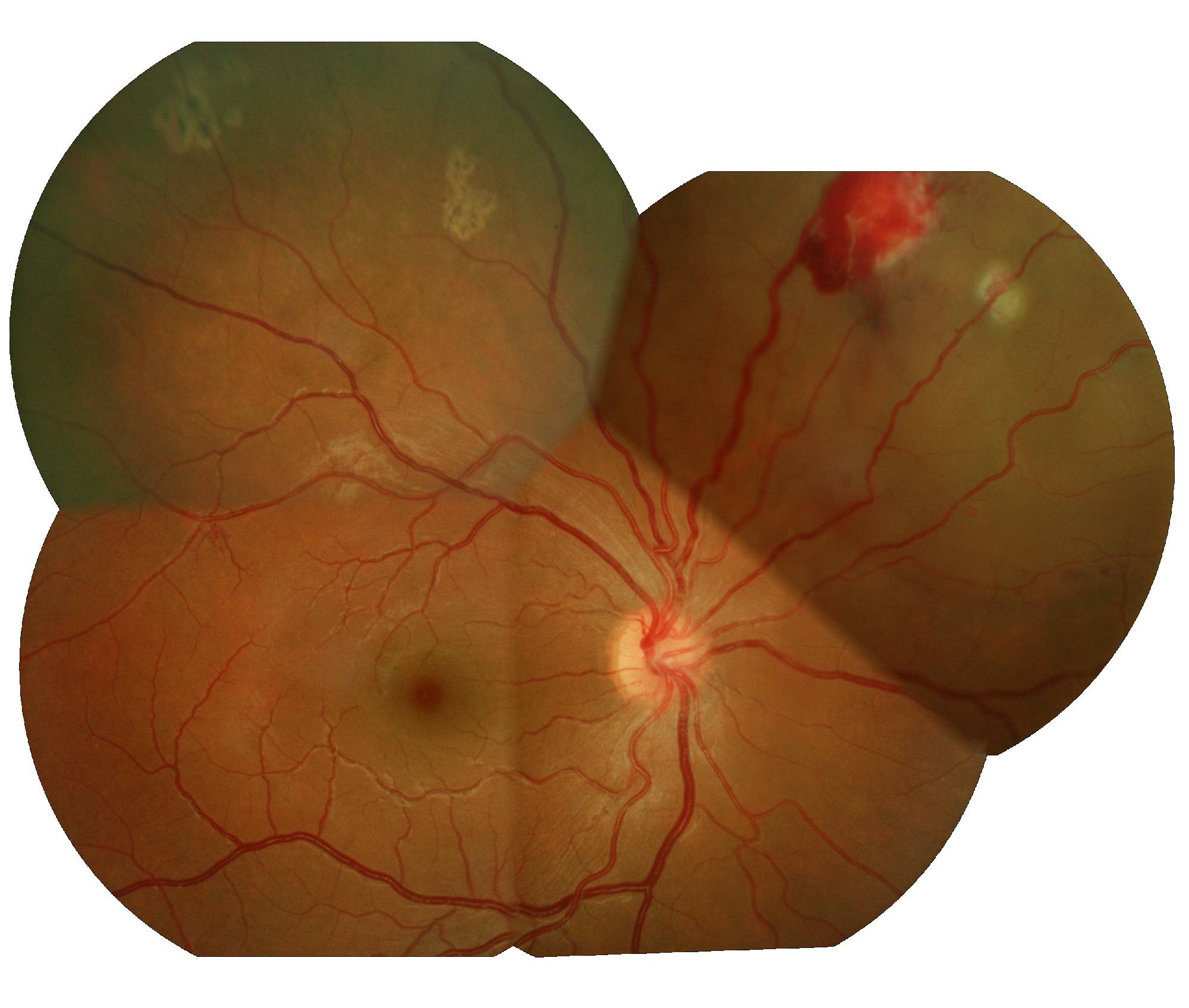 Giovane ragazza con aneurismi retinici multipli in ambo gli occhi trattata pi� volte con argon laser, con un quadro retinico pi� accentuato in OS, dove � presente essudazione con riduzione del campo visivo.
Giovane ragazza con aneurismi retinici multipli in ambo gli occhi trattata pi� volte con argon laser, con un quadro retinico pi� accentuato in OS, dove � presente essudazione con riduzione del campo visivo. Occhio sinistro maggiormente interessato dalla patologia aneurismatica e dove si evidenzia un'essudazione della retina inferiore con soggettiva diminuzioine del campo visivo.
Sindrome di Von Hippel-Lindau
Da Wikipedia, l'enciclopedia libera
Le informazioni qui riportate hanno solo un fine illustrativo: non costituiscono e non provengono n� da prescrizione n� da consiglio medico. Wikipedia non d� consigli medici: leggi le avvertenze.
Sindrome di Von Hippel-Lindau
La VHL o sindrome di Von Hippel-Lindau � una malattia a carattere ereditario molto rara, caratterizzata dall'associazione di diverse forme di neoplasia, fra cui angiomi e altre forme di neoplasia del rene e feocromocitomi.
Indice
[nascondi] 1 Epidemiologia
2 Cenni storici
3 Eziologia
4 Sintomatologia
5 Diagnosi
6 Terapia
7 Bibliografia
8 Voci correlate
9 Altri progetti
Epidemiologia [modifica]
Colpisce una persona su 36.000. L'incidenza � maggiore intorno ai 30-40 anni, anche se pu� manifestarsi a qualunque et�.
Cenni storici [modifica]
Il nome della sindrome � dovuto a coloro che per primi la descrissero: l�oftalmologo tedesco Eugen Von Hippel e il patologo svedese Arvid Lindau.
Eziologia [modifica]
La causa di questa malattia � una mutazione di un gene localizzato sul braccio corto del terzo cromosoma, mappato in 3p25-26, il quale codifica per la proteina VHL. Quest'ultima, normalmente, legandosi ai fattori di trascrizione HIF (Hypoxia Induced Factor) idrossilati dalla presenza di ossigeno, ne provoca l'ubiquitinizzazione e la distruzione tramite il proteasoma. Il complesso coi geni HIF � responsabile delle risposte cellulari all'ipossia, fra cui lo sviluppo di neoangiogenesi e l'induzione della proliferazione cellulare; quindi VHL si comporta come un oncosoppressore e da questo dato sono facilmente deducibili le conseguenze cliniche della sua mancanza. Il prodotto genico pu� essere totalmente assente (delezioni o mutazioni frameshift) con espressione anomala di codoni di STOP) oppure inattivo, portando a quadri clinici differenti.
Sintomatologia [modifica]
A seconda del genotipo del soggetto sono rinvenibili quadri clinici differenti; in particolare la malattia � suddivisibile in 2 tipi. Il tipo 1 con assenza di feocromocitoma e il tipo 2 che invece lo manifesta. Nel primo caso la proteina spesso � mutata, ma presente; nel secondo caso invece � deleta. Inoltre il tipo 2 � diviso ulteriormente in 3 sottotipi: A, B e C (il primo con tumore a cellule renali, il secondo con anche emangioblastoma e il terzo con solo feocromocitoma). Sono anche riscontrati tumori endocrini (specie pancreatici), cisti renali (dovute all'edema da neoangiogenesi) e altre manifestazioni neoplastiche.
Diagnosi [modifica]
La ricerca delle alterazioni del gene VHL deve essere guidata dalla clinica con cui la patologia si presenta e dalla buona pratica clinica. (bisogna valutare l'utilit� reale e la metodica pi� adatta nel fare una consulenza genetica). In particolare nel caso in cui sospetti una delezione dovr� fare gli esami adatti a trovarla, nel caso in cui avr� invece una mutazione puntiforme user� delle metodiche adatte a cercare queste ultime; fatto ci� nel secondo caso dovr� valutare se la mutazione eventualmente trovata corrisponde a patologia; o tramite la consultazione di database appositi oppure facendo delle prove indirette (cerco nei tumori del soggetto anche la delezione dell'allele sano oltre a quello mutato congenitamente, in modo da provare che il danno trovato � responsabile del quadro clinico)
Terapia [modifica]
Si procede ad intervento chirurgico, che viene effettuato in particolari condizioni:
Lesioni spinali sintomatiche
Lesioni cerebrali sintomatiche o in rapida evoluzione
Neoplasie solide renali > 3 cm
Neoplasie solide pancreatiche > 3 cm
Occhio sinistro maggiormente interessato dalla patologia aneurismatica e dove si evidenzia un'essudazione della retina inferiore con soggettiva diminuzioine del campo visivo.
Sindrome di Von Hippel-Lindau
Da Wikipedia, l'enciclopedia libera
Le informazioni qui riportate hanno solo un fine illustrativo: non costituiscono e non provengono n� da prescrizione n� da consiglio medico. Wikipedia non d� consigli medici: leggi le avvertenze.
Sindrome di Von Hippel-Lindau
La VHL o sindrome di Von Hippel-Lindau � una malattia a carattere ereditario molto rara, caratterizzata dall'associazione di diverse forme di neoplasia, fra cui angiomi e altre forme di neoplasia del rene e feocromocitomi.
Indice
[nascondi] 1 Epidemiologia
2 Cenni storici
3 Eziologia
4 Sintomatologia
5 Diagnosi
6 Terapia
7 Bibliografia
8 Voci correlate
9 Altri progetti
Epidemiologia [modifica]
Colpisce una persona su 36.000. L'incidenza � maggiore intorno ai 30-40 anni, anche se pu� manifestarsi a qualunque et�.
Cenni storici [modifica]
Il nome della sindrome � dovuto a coloro che per primi la descrissero: l�oftalmologo tedesco Eugen Von Hippel e il patologo svedese Arvid Lindau.
Eziologia [modifica]
La causa di questa malattia � una mutazione di un gene localizzato sul braccio corto del terzo cromosoma, mappato in 3p25-26, il quale codifica per la proteina VHL. Quest'ultima, normalmente, legandosi ai fattori di trascrizione HIF (Hypoxia Induced Factor) idrossilati dalla presenza di ossigeno, ne provoca l'ubiquitinizzazione e la distruzione tramite il proteasoma. Il complesso coi geni HIF � responsabile delle risposte cellulari all'ipossia, fra cui lo sviluppo di neoangiogenesi e l'induzione della proliferazione cellulare; quindi VHL si comporta come un oncosoppressore e da questo dato sono facilmente deducibili le conseguenze cliniche della sua mancanza. Il prodotto genico pu� essere totalmente assente (delezioni o mutazioni frameshift) con espressione anomala di codoni di STOP) oppure inattivo, portando a quadri clinici differenti.
Sintomatologia [modifica]
A seconda del genotipo del soggetto sono rinvenibili quadri clinici differenti; in particolare la malattia � suddivisibile in 2 tipi. Il tipo 1 con assenza di feocromocitoma e il tipo 2 che invece lo manifesta. Nel primo caso la proteina spesso � mutata, ma presente; nel secondo caso invece � deleta. Inoltre il tipo 2 � diviso ulteriormente in 3 sottotipi: A, B e C (il primo con tumore a cellule renali, il secondo con anche emangioblastoma e il terzo con solo feocromocitoma). Sono anche riscontrati tumori endocrini (specie pancreatici), cisti renali (dovute all'edema da neoangiogenesi) e altre manifestazioni neoplastiche.
Diagnosi [modifica]
La ricerca delle alterazioni del gene VHL deve essere guidata dalla clinica con cui la patologia si presenta e dalla buona pratica clinica. (bisogna valutare l'utilit� reale e la metodica pi� adatta nel fare una consulenza genetica). In particolare nel caso in cui sospetti una delezione dovr� fare gli esami adatti a trovarla, nel caso in cui avr� invece una mutazione puntiforme user� delle metodiche adatte a cercare queste ultime; fatto ci� nel secondo caso dovr� valutare se la mutazione eventualmente trovata corrisponde a patologia; o tramite la consultazione di database appositi oppure facendo delle prove indirette (cerco nei tumori del soggetto anche la delezione dell'allele sano oltre a quello mutato congenitamente, in modo da provare che il danno trovato � responsabile del quadro clinico)
Terapia [modifica]
Si procede ad intervento chirurgico, che viene effettuato in particolari condizioni:
Lesioni spinali sintomatiche
Lesioni cerebrali sintomatiche o in rapida evoluzione
Neoplasie solide renali > 3 cm
Neoplasie solide pancreatiche > 3 cm